Health
Probe of Alzheimer’s follows paths of infection
Starting with microbes, Harvard-MGH researchers outline a devastating chain of events
Sixth in an occasional series on how Harvard researchers are tackling the problematic issues of aging.
What if the bad-boy protein of Alzheimer’s disease — amyloid beta — isn’t so bad after all?
Harvard researchers found themselves asking that question several years ago after noticing remarkable similarities between amyloid beta, thought to be a major player in the disease’s progression, and proteins active in the body’s immune system.
That discovery has blossomed into a new avenue of investigation against the nation’s leading cause of dementia, sixth-deadliest illness, and — according to a 2011 survey — the runner-up to cancer in health fears among the public.
Led by Robert Moir, an assistant professor in neurology at Harvard Medical School (HMS) and Massachusetts General Hospital (MGH), and Rudolph Tanzi, Joseph P. and Rose F. Kennedy Professor of Child Neurology and Mental Retardation at HMS and MGH, the work is focused on whether the development of amyloid beta plaques in the brain — a hallmark of Alzheimer’s disease and the target of several recent drug candidates — might in many cases be a response to infection.
Proven correct, the explanation would fill a significant blank in our framework for the causes of Alzheimer’s disease, create a new understanding of amyloid beta’s role in the body, and possibly open new fronts for treating or preventing the condition by attacking infection before plaques begin to form.
It also has the potential to lump Alzheimer’s with diabetes and other autoimmune diseases in which a revved-up immune system goes too far and turns on the body. Scientists have already noted enough similarities between Alzheimer’s and diabetes that some have wondered whether Alzheimer’s should be thought of as “type 3 diabetes.”
Though knowledge about Alzheimer’s has advanced in recent decades, its causes are only partially understood. In the late 1980s and early 1990s, Tanzi played a role in discovering a trio of genes that cause early onset Alzheimer’s, which runs in a small number of families and can strike before age 50. That condition, also called familial Alzheimer’s, accounts for only about 5 percent of cases.
The remaining cases, called sporadic Alzheimer’s, typically occur later in life, in a person’s mid-60s or beyond. Advancing age is the biggest risk factor — scientists don’t know exactly why — and genetics also plays a role, though less so than in early onset Alzheimer’s.
A variant of the gene APOE, or apolipoprotein E, has been identified as a risk factor in sporadic Alzheimer’s. But having the variant doesn’t make the disease inevitable, and not having it doesn’t rule it out. Which has left scientists wondering what other non-genetic factors are at play.
A significant roadblock to discovery has been a lack of clarity on the role of amyloid beta in the body. The Alzheimer’s community has largely viewed it as an aberrant byproduct that serves no useful purpose in the brain — “metabolic garbage,” as Tanzi once put it.
That view has persisted even as understanding of other aspects of Alzheimer’s has advanced. The prevailing hypothesis today is that amyloid beta aggregates to form plaques in the brain. Those plaques then cause the development of tangles made up of the protein tau within nerve cells. This triggers inflammation — a natural immune response that in this case compounds the damage. Connections between nerve cells are severed and the cells die. Cognitive ability inexorably declines, producing the disease’s most feared outcome.
The idea that Alzheimer’s disease might be caused by infection isn’t new, Moir noted. In fact, in the 1970s and 1980s, some scientists considered it the strongest hypothesis. That changed in 1984 with the discovery of amyloid beta, which came to dominate subsequent research.
“The things creeping around in the brain will scare the heebie-jeebies out of you.”
— Robert Moir
Though support for what came to be called the “pathogen hypothesis” has endured, Tanzi, director of MGH’s Genetics and Aging Research Unit, said that the disease outline he and Moir are developing differs in important ways. While the pathogen hypothesis is most often offered as an alternative to the amyloid beta hypothesis, Moir and Tanzi’s model is not an alternative, but rather fits within the amyloid beta-tau-inflammation paradigm. It fills in blanks, offering an explanation for how the process starts and for the true nature of amyloid beta.
Circumstantial evidence for the importance of amyloid beta is significant, Moir said. It appears to have developed some 400 million years ago and has not only survived evolutionary pressures to appear in humans today, but is present in 60 percent of vertebrates, including fish, reptiles, and birds.
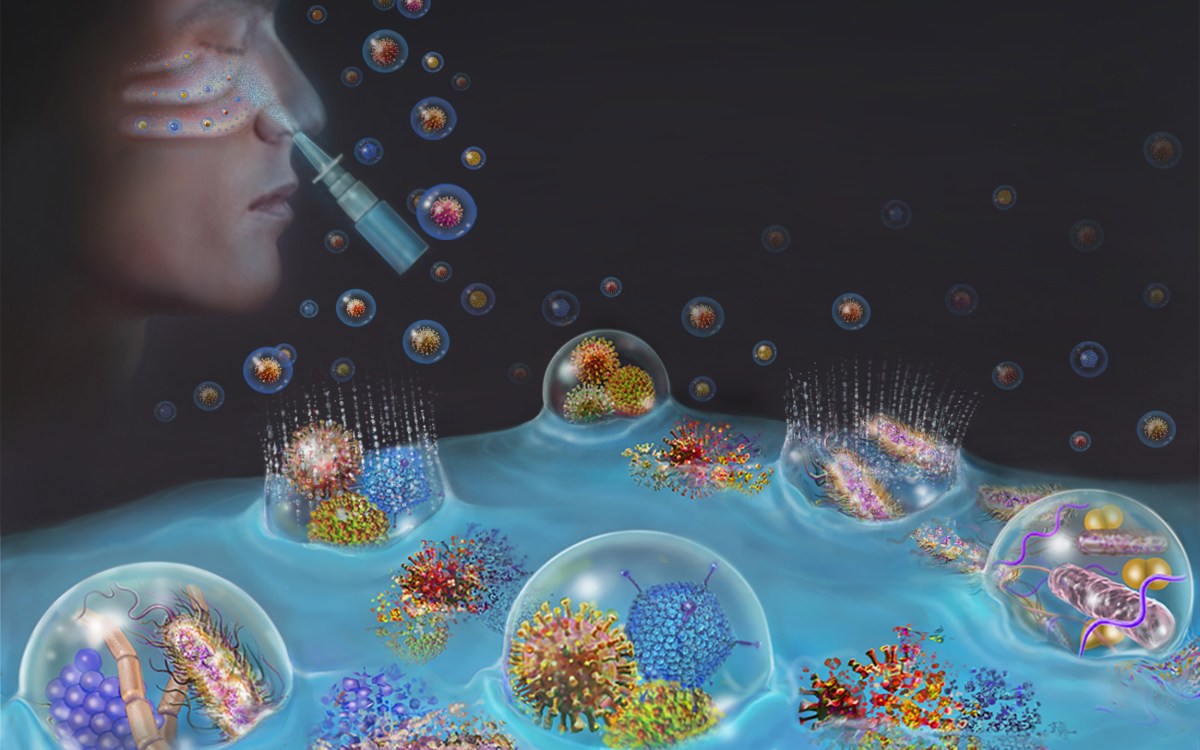
Further, when Moir started to look more closely at the protein, he noticed similarities to key infection-fighting proteins called antimicrobial peptides in the innate immune system — the body’s first and most ancient line of defense.
“It tells you, first, it’s doing something important,” Moir said. “When we started to look at amyloid beta we realized this thing looked similar to antimicrobial peptides.”
Moir compared amyloid beta with LL-37, a potent part of the immune arsenal without which we’d all die from raging infections before reaching our terrible twos. That investigation, supported by a grant from the Cure Alzheimer’s Fund, led to a 2010 paper in which Moir’s team proposed that amyloid beta was an antimicrobial peptide, one that demonstrated infection-fighting abilities comparable to, and in some cases better than, penicillin.
A paper published last year in Science Translational Medicine took the work further, showing that amyloid beta protected against fungal and bacterial infection in nematode worms, laboratory mice, and cell cultures of human neuronal tissue. The work also showed that infecting the brains of lab mice with salmonella resulted in plaque formation 48 hours later, and that those mice lived longer than mice that didn’t develop the plaques.
Alzheimer’s researchers Rudy Tanzi (left) and Robert Moir. Jon Chase/Harvard Staff Photographer
Jon Chase
When the researchers looked even closer, Moir said, at the center of each plaque was a single microbe.
Those findings, backed by the National Institutes of Health, the Cure Alzheimer’s Fund, and the Helmsley Charitable Trust, suggest a model of Alzheimer’s disease initiation that takes into account both infectious and genetic causes, Tanzi said. The process begins with amyloid beta plaques in the brain. In the case of an infection, the culprit is a single microbe — a virus, bacterium, or fungal spore. In the case of genetic disease, seeding relies on a higher-than-normal proportion of extra-long amyloid beta proteins. Amyloid beta, Tanzi explained, comes in two predominant forms. One is a chain of 40 amino acids, which tends to remain in solution, and the other 42 amino acids, which is the predominant form in plaques.
Tanzi compared the plaque formation — whether through amyloid beta’s 42 amino acid form or due to a microbe — to lighting a match. That leads to the development of tau protein tangles in nerve cells, which Tanzi compared to a brush fire. Then comes inflammation, a raging forest fire which Tanzi believes does much of the damage that leads to cognitive decline.
“It’s inflammation that really throws you down the slippery slope,” he said.
The brain was once thought of as kept relatively sterile by the blood-brain barrier, a membrane that selectively admits key molecules such as glucose and amino acids while blocking potentially harmful invaders.
Some notable diseases have long been known to slip through, including syphilis and rabies. But recently it’s become apparent that there’s a lot more life in there than scientists had understood, Moir said. “The things creeping around in the brain will scare the heebie-jeebies out of you.”
Figuring out what is and isn’t in the brains of Alzheimer’s patients is the logical next step, one that Moir and Tanzi are undertaking through the Brain Microbiome Project. The researchers are examining brain bank samples from people who died of Alzheimer’s with those who didn’t. In the initial phase, they are searching for viruses by scanning for their genetic material.
When all is said and done, Moir said, it might be that Alzheimer’s is a “dysbiosis” of the brain, a sign that the microbiome is out of whack. Further, Tanzi said, if microbial culprits are identified, it might be possible to treat those infections early in life — possibly even with a vaccine — before the earliest plaques begin to form.
“What you see is a lot of stuff,” Moir said. “A lot of scary stuff.”